Welcome!
We are a computational research group at the Institute for Computational Cancer Biology Cologne (ICCB) at the University of Cologne and the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at the TU Berlin.
Cancer genomics and evolution
Our lab develops and applies algorithms and computational methods to understand how cellular and intra-tumour heterogeneity (ITH) arises and how it affects tissue and patient phenotypes in space and time. We are particularly interested in chromosomal instability (CIN) and somatic copy-number alterations (SCNA), a key characteristic that separates cancerous from healthy somatic tissue. In our methods we leverage statistical and machine learning approaches as well as classical computer science algorithms and simulations and develop these models in close collaboration with our experimental partners.
Specifically, we are active in three research areas:
- Structural evolution of cancer genomes. We infer cancer evolution from clinical patient samples and develop forward simulations that enable us to investigate cancer growth and evolution in-silico. These advances will help us one day to predict cancer evolution and provide treatment suggestions that avoid resistance. #medicc #refphase #smith
- Haplotyping and allele-specific effects of genetic variation. We develop algorithms to reconstruct haplotypes from sequencing data and apply machine learning and statistical genetics approaches on large patient cohorts to understand cancer gene regulation and epigenetics. #gamibhear #refphase
- Cancer early detection and prevention. We develop machine learning methods to identify and exploit population-based molecular biomarkers for risk screening and stratification, as well as for prognosis of outcome and relapse. #mop-c
Our work bridges theoretical and applied biomedical research and we develop, train, and validate our methods on large clinical datasets as part of the Pan-Cancer Analysis of Whole Genomes (PCAWG), ICGC-ARGO, and the TRACERx Consortium as well as several smaller consortia.
Locations


Our main location is at the University Hospital Cologne in the vibrant, international city of Cologne, Germany.
We are also maintaining a branch at the Berlin Institute for the Foundations of Learning and Data (BIFOLD) in the German capital of Berlin.
Keep an eye on our recruitment page for job openings at either location.
April 2, 2024
News & Views in Nature Genetics
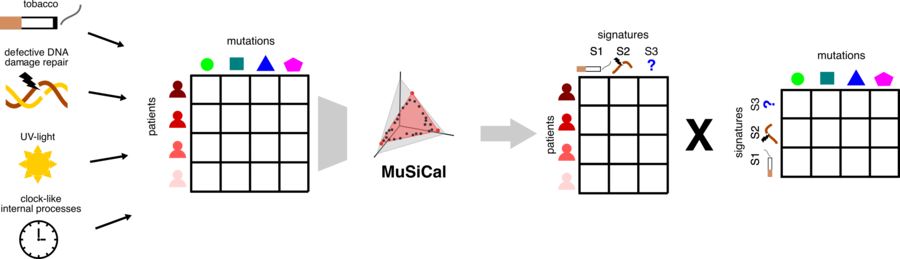
Roland and Tom were invited to give their views on the Nature Genetics paper of Jin et al. presenting MuSiCal, a set of novel algorithms for improved identification of cancer mutational processes by mutational signatures.
October 23, 2023
Refphase paper published in PLOS Computational Biology
We are excited that after 9 years of development, our multi-region copy-number phasing algorithm Refphase is finally here!
Refphase uses WES or WGS data from multiple samples from the same patients to identify the haplotypes of origin of somatic copy-number alterations (SCNAs). Refphase has led to the discovery of the Mirrored Subclonal Allelic Imbalance (MSAI) phenomenon in cancer (Jamal-Hanjani et al. 2017, Nature), where SCNAs affect opposite haplotypes in the same tumour, leading to a "mirrored" B-allele frequency pattern.
Refphase was further key to demonstrating widespread MSAI occurrences and continuous parallel evolution across human cancers (Watkins et al. 2020, Nature).
Refphase is implemented in R and available on Bitbucket at https://bitbucket.org/schwarzlab/refphase.
May 18, 2023
Marina Petkovic successfully defends her PhD

We are thrilled to report the third successful PhD defense in the Schwarzlab: Marina Petkovic defended her PhD on May 12, 2023 at the Berlin Institute for Molecular Systems Biology, Max Delbrück Center for Molecular Medicine in the Helmholtz Association.
Her PhD thesis is titled "Reconstructing the evolutionary history of cancer from allele-specific copynumber profiles" and she will receive her PhD from the Humboldt University Berlin.
During her PhD she contributed in a major way to the development of MEDICC2 and conducted large scale analyses on the order of evolutionary copy-number events in cancer.
After an interim postdoc at Charite Berlin, Marina is now moving to greener pastures in industry where she will continue to work in bioinformatics.
Research
Publications
Key publications
TBK Watkins, EC Colliver, MR Huska, TL Kaufmann, ..., McGranahan N, Schwarz RF. Refphase: Multi-sample phasing reveals haplotype-specific copy number heterogeneity. PLOS Computational Biology (2023).
▶ Refphase uses genomic regions of allelic imbalance across multiple samples from the same patient to phase germline variants and somatic copy-number alterations.
Streck A, Kaufmann T, Schwarz RF. SMITH: Spatially Constrained Stochastic Model for Simulation of Intra-Tumour Heterogeneity Bioinformatics (2023).
▶ SMITH is a novel method for simulating cancer evolution to realistic tumour sizes of more than one billion cells that also models spatial constraints. We found an acronym! That was close.
Kaufmann T, Petkovic M, Watkins TBK, ..., Van Loo P, Haase K, Tarabichi M, Schwarz RF. MEDICC2: whole-genome doubling-aware copy number phylogenies for cancer evolution. Genome Biology (2022).
▶ MEDICC2 is the leading method for inferring cancer evolution from somatic copy-number alterations. It identifies individual evolutionary events and detects whole-genome doubling. Published only eight years after MEDICC! Second best acronym ever.
Markowski J, Kempfer R, ... , Kehr B, Pombo A, Rahmann S, Schwarz RF. GAMIBHEAR: whole-genome haplotype reconstruction from Genome Architecture Mapping data. Bioinformatics (2021).
▶ GAMIBHEAR is a novel algorithm for inferring chromosome-spanning haplotypes from Genome Architecture Mapping data. It provides the basis for accurate haplotype-specific chromatin contact maps in human. Best acronym ever.
PCAWG Transcriptome Core Group, Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, ...,
Brazma A*, Brooks A*, Göke J*, Rätsch G*, Schwarz RF*, Stegle O*, Zhang Z*. Genomic basis for RNA alterations in cancer. Nature (2020).
▶ In the PCAWG consortium we investigated the allele-specific effects of somatic mutations on gene expression as part of PCAWG Working Group 3. Another interesting acronym story.
Watkins TBK, Lim EL, Petkovic M, Elizalde S, Birkbak NJ, Wilson GA, Moore DA, ..., Schwarz RF*, McGranahan N*,
Swanton C*. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature (2020).
▶ In this seminal paper we used the reference phasing algorithm we developed to detect parallel evolution across human cancers in the largest multi-region sequencing dataset to date. Go refphase!
Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, ..., Schwarz RF, et al.
Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med., 376(22):2109–2121 (2017).
▶ In this work, we developed and contributed the prototype of the refphase phasing algorithm to the TRACERx consortium, which lead to the detection of mirrored subclonal allelic imbalance events (MSAI). #notmyacronym
People
Meet the Team

Group Leader
Prof. Dr. Roland F. Schwarz
Computer scientist by training. Lover of formal grammars, Markov models and phylogenetic trees.
Quote: "What if it's a Markov chain?"

PhD Student
Maja-Celine Stöber
Biomathematician. Fond of single-cell data analysis and extrachromosomal DNA. Master of Journal Club.
Quote: "No, Roland, you cannot skip Journal Club."


Postdoc / Scientific Programmer
Dr. Adam Streck
Computer scientist by training. Modelling the world through cellular automata and stochastic processes. Gamer at heart, living the VR hype. Author of SMITH. Master of Technology.

Clinician Scientist
Dr. Daniel Schütte
Medical doctor and computer scientist. Improving patient care through early detection and better stratification.

MD Student
Felix Schifferdecker
Medical student and computer scientist. Simulating cancer evolution and structural alterations.

MD Student
Selina Wächter
Medical student. Investigating how selectional constraints shape cancer evolution.

Postdoc
Dr. Cody Duncan
Physicist by training. Interested in simulation-building and stochastic processes. In-house Rugby League expert. Master of Technology Cologne.

Postdoc
Dr. Nathan Lee
Applied mathematician & computational biologist. Interested in cancer evolution, stochastic processes, and simulations of carcinogenesis.

PhD Student
Claudia Robens
Biotechnologist by training. Investigating chromosomal instability and chromatin architecture in cancer.

PhD Student
Katyayni Ganesan
Biologist by training. Interested in single-cell cancer evolution and transcriptomics.

Postdoc / Scientific Coordinator
Dr. Laura Godfrey
Biologist by training. Interested in epigenetics and -genomics.
Scientific coordinator, third party funding, web editor.

Master Student
Giuseppe Barranco
Biologist and bioinformatician by training. Interested in single-cell genomics.
PhD Student
Chenxi Nie
Bioinformatician by training. Interested in stochastic processes, stochastic sampling and Formula 1.

Administrative Assistant
Stefanie Fleer
Lab coordination and administrative organisation
Consortium
CRUK - TRACERx
We are part of the CRUK funded TRACERx consortium lead by Charles Swanton at the Francis Crick Institute in London where we contribute algorithms for phasing of copy-number alterations and phylogenetic tree inference.
Consortium
ICGC - ARGO
In ICGC-ARGO we are part of the data coordination and management group and are leading a project that contributes pipelines for allele-specific expression analysis.